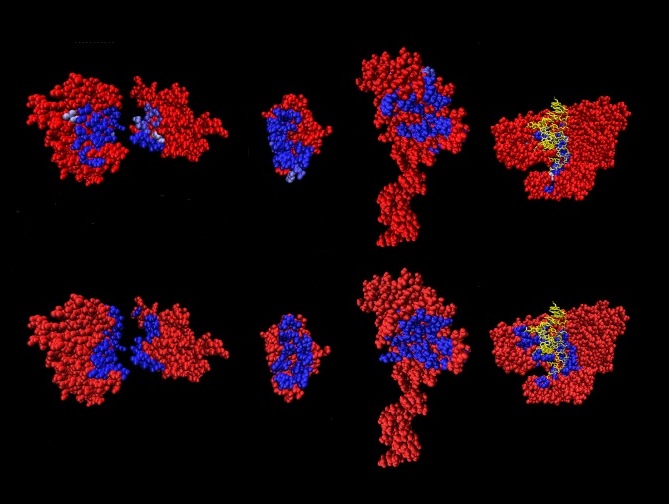

Neuromuscular disease is a generic term for a group of
disorders (more than 200 in all) that impair muscle functioning
either directly through muscle damage (muscular dystrophy) or
indirectly damaging nerves. It affects one in 2,000 people. These
chronic diseases lead to a decrease in muscle strength, causing
serious disabilities in motor functions (moving, breathing etc.).
The most well-known is muscular dystrophy. In cases of muscular
dystrophy contraction of the muscle leads to disruption of the
outer membrane of the muscle cells and eventual weakening and
wasting of the muscle. Dystrophin is part of a protein complex
that connects the cytoskeleton of a muscle fiber to the tissue
framework surrounding each cell through the cell membrane. This
complex does not form correctly in muscular dystrophy. (Image
courtesy Alessandra Carbone).
Proteins are part of a complex social network, and rarely act alone. Protein-protein interactions is the term used to describe when two or more proteins 'partner-up' and bind together to carry out a different biological function. While are used to identify the relationships between one protein and another in its cellular neighborhood, computational simulations are still needed to uncover the more complex web of connections for multiple protein partners.
Distributed computing power from the has recently aided the project in capturing all the possible molecular and atomic connections between 2,280 human proteins. The analyzed proteins include those that are known to mutate and induce different forms of neuromuscular disorders, including Muscular Dystrophy.
HCMD is part of a larger-scale venture, . This is an alliance between AFM (French Muscular Dystrophy Association), CNRS (French National Center for Scientific Research) and IBM, who are using the World Community Grid resources to help them decipher and map all the functions of interacting proteins found in humans to a worldwide repository of information such as the Research Collaboratory for Structural Bioinformatics (RCSB) .
The idea behind molecular docking simulations is to take two proteins from a database of proteins (of known structure), and to see which proteins have an affinity to bind to one another. This involves predicting the position and orientation (the 3D-structure) of a protein in relation to a (another protein, DNA, drug, etc.).
"You take two proteins and see in space how they best fit rotating one protein around the sphere of another," says Alessandra Carbone from the Pierre-and-Marie-Curie University Paris, who is lead researcher on the project. "There might be 300,000 possible positions for two given proteins to test. The number of configurations varies according to the size of the proteins. This means for larger proteins there can be over 300,000 positions. Each one of these calculations can take 30 seconds so the calculations for two proteins can take almost a month on a PC." This fit is also dependent on both geometrics (how well the surface shapes complement each other) and the chemical bonds (measuring the strength of the atomic interactions between the two molecules).
The first phase of the HMCD project, completed in June 2007, scrutinized relationships among 168 proteins using molecular docking simulations. "Humans have more than 20,000 proteins, so if we were to examine the full range, it would be a truly mammoth task and we would have centuries and centuries of calculations. Even just a hundred proteins will give you centuries of calculations if you explore all 300,000 configurations for each protein. Molecular modeling simulations compute the energy values at the atomic level for groups of atoms between the given proteins. It gives you some sort of numerical estimation on whether two proteins are likely to interact or not, or likely to repulse each other depending on the physical or chemical properties of the amino acids on the surface, once you combine it with knowledge of where the interaction site lies," says Carbone.
The researchers predicted that it would have taken over 14,000 years of computational time on a 2 GHz PC to reveal and rule out all possible docking confirmations for all 168 proteins. However, a 'distributed calculation' allowed them to considerably reduce the processing time. Over 6,000-8,000 donor machines meant the task took under 26 weeks.
However, to test 2,280 proteins on a one-to-one basis for phase II of the project, researchers needed a method to significantly reduce the number of configurations they would have to check. Molecular docking data from analysis of the 168 proteins (known to form 84 complexes) helped them develop a fast docking algorithm to predict potential partners for this large pool of proteins.
The researchers also combined evolutionary information with these molecular docking algorithms. Cells across all species have a similar way of organizing their internal structures with many protein architectures being conserved (or homologous) across species. Protein-protein interactions from one organism can, therefore, be used to predict interactions among homologous proteins in another organism. The interaction sites on the protein surface are usually conserved because the amino acids or physical properties are conserved. "Evolutionary information lets us predict the interaction site (binding site) with reasonable probability, and then we can carry out molecular docking just on this area which decreases exploration time even further. While you usually explore the whole sphere around a given protein, evolutionary analysis allowed us to explore just a small part of the surface area or cross section - a cone of the sphere," says Carbone. "The numerical estimation to discriminate proteins is realized by combining the molecular docking results and the predictions of the binding sites based on evolution," she adds.
The project came to an end ahead of schedule in September 2012 after only two and half years. Volunteer computers had racked up over 114 million results using nearly 53,000 CPU-years of computing power.
HCMD2 expect to publish their results, methods, scientific tools and interaction data in a number of public databases, along with descriptive papers, later this year. The resulting database will potentially help drug developers design molecules to inhibit or enhance binding of particular macromolecules, hopefully leading to better treatments for muscular dystrophy and other neuromuscular diseases. The unique methodology used by the researchers in their analysis could also be applied to future studies of other disease areas.
{kind=link}