
- Large-scale protein studies are swamped under large data sets.
- What if deductions could isolate only the relevant data?
- MS-REDUCE algorithm speeds analyses and paves the way for precision medicine.
The data tsunami has finally come ashore in the world of protein research. Not to worry — US -funded research out of has procured a lifeboat and with the help of high-performance computing (HPC) has sped up analysis 100-fold. Next on the horizon: precision medicine for your grandmother.
A is a tool used to measure the molecular mass of a given biological sample. In the world of modern large-scale protein study (), a MS can generate huge waves of data in a short time. Each sample spectrum will include thousands of data peaks that scientists must wade through to determine a peptide sequence.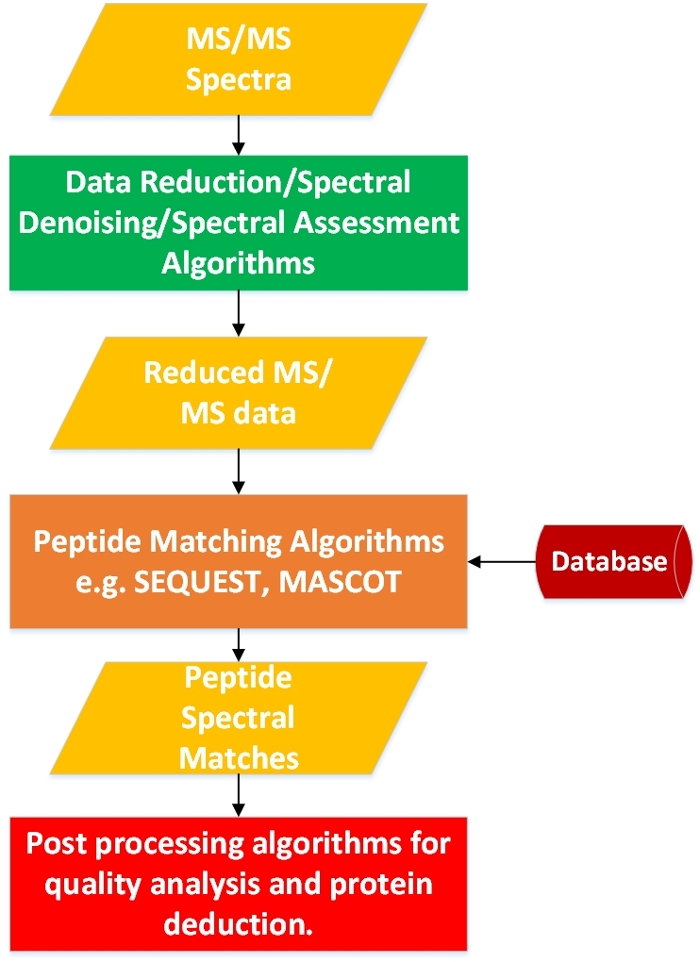
“The assumption that all the proteomic tools until now have worked under is that all the data is relevant,” says , computer science professor and director of the Parallel Computing and Data Science (PCDS) lab at Western Michigan University. “We say this assumption is incorrect, and maybe we can come up with techniques to allow us to take only the relevant data out of those big data sets — and maybe some quicker deductions are possible.”
Assuming all proteomic data is relevant wouldn’t be so bad, but as mass spectrometers have become more sophisticated, data sets mount at a rapid rate. A moderate-sized proteomic facility can produce terabytes of data in a few hours, and a petabyte in a few weeks. Storage and analysis quickly founder in this deluge. Until recently, researchers were drowning in this bottleneck, mired in inefficient computational techniques. But in research , Saeed’s team has capsized conventional assumptions about the value of every segment of data churned up by mass spectrometers.
"Our research is helping to build the foundation needed to reach personal medicine in the coming years." ~ Fahad Saeed.
Other noise reduction tools have attempted to ford this stream of data without much success, but MS-REDUCE, Saeed’s breakthrough reduction algorithm, is demonstrating analyses 100 times faster than existing noise-removing algorithms.
“All of these previous approaches have been focused on noise reduction, but we are going one step ahead of them, saying, ok it’s not only the noise we want to reduce, but we want to reduce all the data points that are not relevant,” says Saeed.
Saeed has benchmarked the MS-REDUCE algorithm on a moderate high-performance computing server and produced some impressive results. Comparable reduction tools required three days to process 1 million spectra — MS-REDUCE finished in less than one hour.
 WMU’s modest HPC environment was ideal for Saeed’s ultimate purpose of making the technology accessible to practitioners. Though proteomic data sets require HPC architecture for analysis, Saeed's goal is to make the process more manageable and more affordable so our grandmothers need not drive to the nearest supercomputing center.
WMU’s modest HPC environment was ideal for Saeed’s ultimate purpose of making the technology accessible to practitioners. Though proteomic data sets require HPC architecture for analysis, Saeed's goal is to make the process more manageable and more affordable so our grandmothers need not drive to the nearest supercomputing center.
“At some point in her life, my grandmother is going to go to the hospital and order a blood test,” Saeed observes. “We are working toward the day that she will be able to get her genome and proteome analyzed — quickly — and see if there are any deficiencies or problems that can be dealt with. Our research is helping to build the foundation needed to reach personal medicine in the coming years.”
At its core, Saeed’s work is another NSF-investment success story. He credits the for the construction and continual maintenance of the PCDS lab at WMU, and for the support for students and junior scholars in his program.
“I was blessed with mentors who motivated me to do this kind of work, so we encourage K-12 and undergraduate students to interact with me, my PhD students, and other researchers in the lab. I’m more than happy and very excited to do this so that the next generation of scientists can do groundbreaking work.”
{kind=link}